الأنيميا البحرية / الثالاسميا
سجية الأنيميا البحرية
Thalassemia
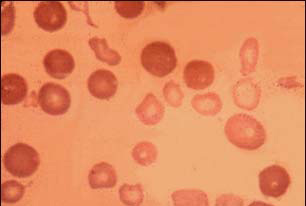
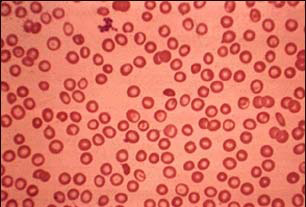
الثالاسيميا أو الأنيميا البحرية هي اضطراب وراثي يصيب خلايا الدم الحمر، سببه
عيب في عملية إنتاج الهيموجلوبين (وهو البروتين الحامل للاكسجين في تلك الخلايا).
في الصور الشديدة من المرض تكون خلايا الدم الحمر التي ينتجها نخاع العظام هشة
وسهلة التحطم، مما يسبب أنيميا التحلل الدموي.
الثالاسيميا (ويسمى أحد أشكالها أنيميا البحر المتوسط) تحدث في أكثر الحالات بين
الشعوب ذوي الأصول المنتمية إلى إيطاليا واليونان والشرق الأوسط وجنوب اسيا
وأفريقيا. ينتقل المرض عن طريق الآباء الذين يحملون جين الثالاسيميا. وحامل المرض
يكون لديه واحد أو أكثر من الجينات الطبيعية، وهي الحالة التي تعرف بالصورة الطفيفة
من الثالاسيميا (أو ما يسمى سجية أنيميا البحر المتوسط Thalassemia Trait).
أغلب من لديهم تلك الحالة يعيشون حياة طبيعية صحية (أي أنه حامل للمرض فقط ولكنه
غير مريض).
لكن عندما يتزاوج حاملان للمرض ويصبحان أبوين يكون هناك احتمال بنسبة واحد من
أربعة أن يرث الطفل الوليد جينا للثالاسيميا من كل من أبويه فيصاب بصورة شديدة من
المرض. وهناك احتمال بنسبة اثنين من أربعة أن يرث الطفل جيناً طبيعيا واخر غير
طبيعي فيصبح حاملا للمرض مثل أبويه. وهناك احتمال بنسبة واحد من أربعة أن يرث
الطفل جينين طبيعيين من أبويه فيصبح خاليا تماما من المرض ومن حالة حمل المرض
كذلك.
قد يفضل الأبوان أن يحصلا على استشارة جينية لتحديد ما إذا كانا يحملان الجين
الوراثي للثالاسيميا. اختبار ما قبل الولادة بأخذ عينات من الخملات المشيمية أو
بزل السائل الأمنيوسي يمكن أن يكشف عن الثلاسيميا في الجنين.
الصورتان الرئيسيتان من الثلاسيميا تسميان ألفا ثلاسيميا ALPHA THALASSEMIA و
بيتا ثالاسيميا BETA THALASSEMIA، ويعتمد هذا التقسيم على الجزء من الهيموجلوبين
الذي تأثر بالحالة المرضية في خلايا الدم الحمر.
أما الفا – ثالاسيميا فنادرة. والغياب التام لألفا هيموجلوبين يؤدي دائما إلى موت
الجنين أو حديث الولادة. الصورة الطفيفة (أو حمل المرض) من نوع ألفا تكون شائعة
بوضوح، ولكن نادرا ما تؤدي إلى مشاكل خطرة. يمكن أن تؤدي فقط إلى انيميا طفيفة
تكون فيها خلايا الدم الحمر أصغر حجما قليلا من الخلايا الطبيعية.
أما بيتا ثلاسيميا وتسمى أيضا أنيميا البحر المتوسط فتتزاوج بين الصور التي لا
تؤثر مطلقا على الصحة إلى الصور الشديدة جدا. يمكن أن يشخص الطبيب حالة بيتا
ثالاسيميا عن طريق مسحات الدم التي تكشف خلايا الدم الحمر المميزة بحجمها الصغير
ولونها الباهت، وعن طريق اختبار الدم الذي يسمى الانتقال الكهربائي للهيموجلوبين.
التشخيص المبكر للصورة الشديدة من التلاسيميا أساسي لمنع أكبر عدد ممكن من
المضاعفات. في هذه الثلاسيميا الشديدة سرعان ما يصاب الطحال والكبد والقلب بالتضخم
الشديد ما لم تعالج الحالة. كما تصبح العظام رقيقة وهشة وتصبح عظام الوجه مشوهة.
ويكون هبوط القلب والإصابة بالعدوى هما السببان الرئيسيان لموت الأطفال الذين لم
يعالجوا من تلك الحالة.
أغلب الأطفال المصابين بحالة بيتا ثالاسيميا يبدون أصحاء عند الولادة، ولكن أثناء
الستة أشهر الأولى يكونون ضعفاء وسريعي الهياج وشهيتهم ضعيفة والطحال لديهم متضخم
بسبب التحطيم الزائد لخلايا الدم الحمر. تنمو العظام بشكل غير طبيعي كلما كبر نخاع
العظام وتمدد لتعويض العمر القصير لخلايا الدم الحمر. ويحدث غالباً اصفرار للجلد
ويسمى اليرقان.
عندما يعالج الأطفال المصابون بالثالاسيميا بنقل متكرر للدم (بصفة عامة مرة كل 3
إلى 4 أسابيع) وذلك بهدف المحافظة على مستوى الهيموجلوبين لديهم قريبا من
الطبيعي، يمكن تجنب كثير من المضاعفات. مع ذلك، فإن تعدد وتكرار نقل الدم يؤدي إلى
تراكم الحديد في الجسم مما قد يؤدي إلى إلحاق الضرر بالقلب والكبد وأعضاء أخرى. لذا
يراعى إعطاء دواء مقتنص للحديد للمساعدة على تخليص الجسم من الحديد الزائد عن
الحاجة، وبهذا يمكن منع أو تأخير المشكلات المتعلقة بزيادة كميات الحديد. يعطى
العقار عادة عن طريق مضخة آلية تضخ العقار تحت الجلد بينما يكون الطفل نائماً.
الأطفال المرضى بالثلاسيميا الشديدة الذين يعالجون بنقل الدم المتكرر مع اقتناص
الحديد الزائد يمكنهم الحياة لمدة عشرين إلى ثلاثين سنة أو أكثر. قد أمكن شفاء
الثالاسيميا باستخدام طريقة زرع نخاع العظام. مع ذلك فإن هذا النوع من العلاج
متاح فقط لنسبة قليلة من الناس الذين يتاح لهم وجود متبرع مناسب لنخاع العظام. هذا
علاوة على أن تقنية الزرع تكون محفوفة بالمخاطر ويمكن أن تؤدي إلى الوفاة.